



Table of Contents
The Telomere-to-Telomere Consortium is sequencing whole chromosomes.Credit: Adrian T. Sumner/SPL
From gene editing to protein-structure determination to quantum computing, here are seven technologies that are likely to have an impact on science in the year ahead.
Fully finished genomes
Roughly one-tenth of the human genome remained uncharted when genomics researchers Karen Miga at the University of California, Santa Cruz, and Adam Phillippy at the National Human Genome Research Institute in Bethesda, Maryland, launched the Telomere-to-Telomere (T2T) consortium in 2019. Now, that number has dropped to zero. In a preprint published in May last year, the consortium reported the first end-to-end sequence of the human genome, adding nearly 200 million new base pairs to the widely used human consensus genome sequence known as GRCh38, and writing the final chapter of the Human Genome Project1.
First released in 2013, GRCh38 has been a valuable tool — a scaffold on which to map sequencing reads. But it’s riddled with holes. This is largely because the widely used sequencing technology developed by Illumina, in San Diego, California, produces reads that are accurate, but short. They are not long enough to unambiguously map highly repetitive genomic sequences, including the telomeres that cap chromosome ends and the centromeres that coordinate the partitioning of newly replicated DNA during cell division.
Long-read sequencing technologies proved to be the game-changer. Developed by Pacific Biosciences in Menlo Park, California, and Oxford Nanopore Technologies (ONT) in Oxford, UK, these technologies can sequence tens or even hundreds of thousands of bases in a single read, but — at least at the outset — not without errors. By the time the T2T team reconstructed2,3 their first individual chromosomes — X and 8 — in 2020, however, Pacific Biosciences’ sequencing had advanced to the extent that T2T scientists could detect tiny variations in long stretches of repeated sequences. These subtle ‘fingerprints’ made long repetitive chromosome segments tractable, and the rest of the genome quickly fell into line. The ONT platform also captures many modifications to DNA that modulate gene expression, and T2T was able to map these ‘epigenetic tags’ genome-wide as well4.
The genome T2T solved was from a cell line that contains two identical sets of chromosomes. Normal diploid human genomes contain two versions of each chromosome, and researchers are now working on ‘phasing’ strategies that can confidently assign each sequence to the appropriate chromosome copy. “We’re already getting some pretty phenomenal phased assemblies,” says Miga.
This diploid assembly work is being conducted in collaboration with T2T’s partner organization, the Human Pangenome Reference Consortium, which aspires to produce a more representative genome map, based on hundreds of donors from around the world. “We’re aiming to capture an average of 97% of human allelic diversity,” says Erich Jarvis, one of the consortium’s lead investigators and a geneticist at the Rockefeller University in New York City. As chair of the Vertebrate Genomes Project, Jarvis also hopes to leverage these complete genome assembly capabilities to generate full sequences for every vertebrate species on Earth. “I think within the next 10 years, we’re going to be doing telomere-to-telomere genomes routinely,” he says.
Protein structure solutions
Structure dictates function. But it can be hard to measure. Major experimental and computational advances in the past two years have given researchers complementary tools for determining protein structures with unprecedented speed and resolution.
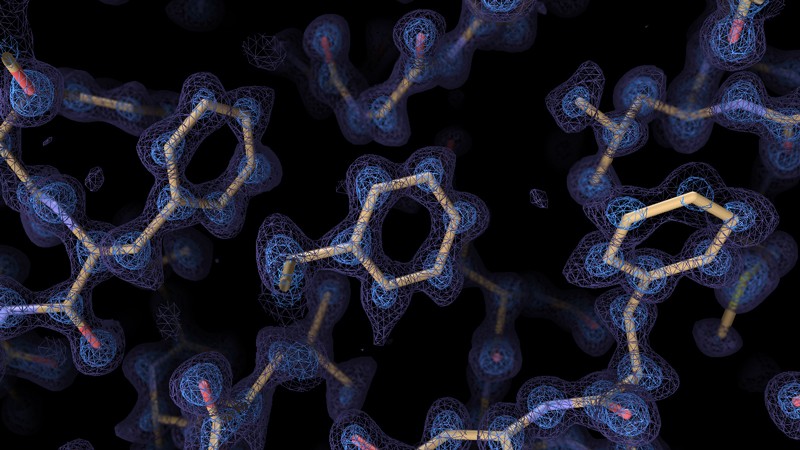
The AlphaFold2 structure-prediction algorithm, developed by Alphabet subsidiary DeepMind in London, relies on ‘deep learning’ strategies to extrapolate the shape of a folded protein from its amino acid sequence5. Following a decisive victory at the 2020 Critical Assessment of protein Structure Prediction competition, in which computational biologists test their structure-prediction algorithms head-to-head, AlphaFold2’s reputation — and adoption — has soared. “For some of the structures, the predictions are almost eerily good,” says Janet Thornton, senior scientist and former director of the European Bioinformatics Institute in Hinxton, UK. Since its public release last July, AlphaFold2 has been applied to proteomes, to determine the structures of all the proteins expressed in humans6 and in 20 model organisms (see Nature 595, 635; 2021), as well as nearly 440,000 proteins in the Swiss-Prot database, greatly increasing the number of proteins for which high-confidence modelling data are available. The AlphaFold algorithm has also proven its ability to tackle multi-chain protein complexes7.
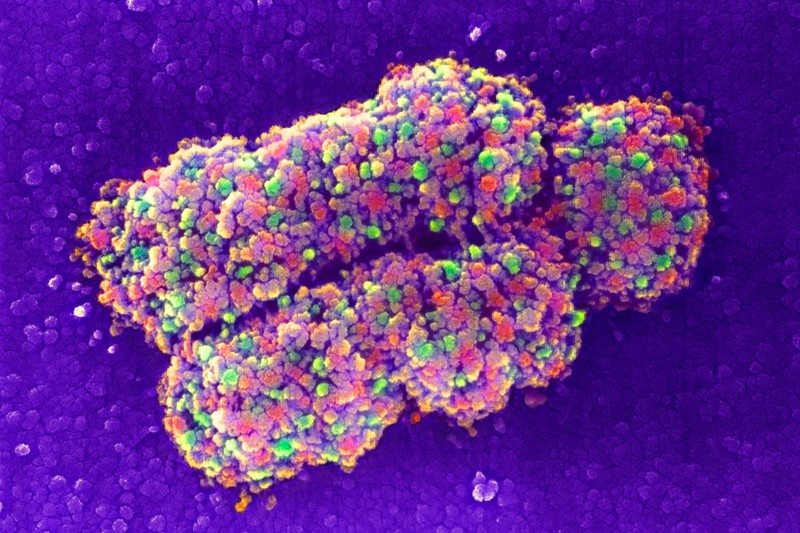
In parallel, improvements in cryogenic-electron microscopy (cryo-EM) are enabling researchers to experimentally solve even the most challenging proteins and complexes. Cryo-EM scans flash-frozen molecules with an electron beam, generating images of the proteins in multiple orientations that can then be computationally reassembled into a 3D structure. In 2020, improvements in cryo-EM hardware and software enabled two teams to generate structures with a resolution of less than 1.5 ångströms, capturing the position of individual atoms8,9. “Prior to this, we bandied about the term ‘atomic resolution’ with wild abandon, but it’s only been near-atomic,” says Bridget Carragher, co-director of the New York Structural Biology Center’s Simons Electron Microscopy Center in New York City. “This truly is atomic.” And, although both teams used an especially well-studied model protein called apoferritin, Carragher says, these studies suggest that near-atomic resolution is feasible for other, more difficult targets as well.
Images from cryo-electron microscopy are helping to solve complex structures.Credit: Paul Emsley/MRC Laboratory of Molecular Biology
Many experimentalists who were initially sceptical of AlphaFold2 now see it as a clear complement to experimental methods such as cryo-EM, where its computational models can aid in data analysis and reconstruction. And cryo-EM can generate findings currently out of reach for computational prediction. Carragher’s team, for instance, is using ‘time-resolved’ cryo-EM to capture rapid conformational changes that occur when proteins interact with other molecules. “We can trap things and see what’s happening on the order of a hundred milliseconds,” she says.
There is also considerable excitement around a related method, cryo-electron tomography (cryo-ET), which captures naturalistic protein behaviour in thin sections of frozen cells. But interpretation of these crowded, complicated images is challenging, and Carragher thinks computational advances from the machine-learning world will be essential. “How else are we going to solve these almost intractable problems?” she asks.
Quantum simulation
Atoms are, well, atomic in size. But under the right conditions, they can be coaxed into a highly-excited, super-sized state with diameters on the order of one micrometre or more. By performing this excitation on carefully positioned arrays of hundreds of atoms in a controlled fashion, physicists have demonstrated that they can solve challenging physics problems that push conventional computers to their limits.
Quantum computers manage data in the form of qubits. Coupled together using the quantum physics phenomenon called entanglement, qubits can influence each other at a distance. These qubits can drastically increase the computing power that can be achieved with a given allotment of qubits relative to an equivalent number of bits in a classical computer.
Several groups have successfully used individual ions as qubits, but their electrical charges make them challenging to assemble at high density. Physicists including Antoine Browaeys at the French national research agency CNRS in Paris and Mikhail Lukin at Harvard University in Cambridge, Massachusetts, are exploring an alternative approach. The teams use optical tweezers to precisely position uncharged atoms in tightly packed 2D and 3D arrays, then apply lasers to excite these particles into large-diameter ‘Rydberg atoms’ that become entangled with their neighbours10,11. “Rydberg atom systems are individually controllable, and their interactions can be turned on and off,” explains physicist Jaewook Ahn at the Korea Advanced Institute of Science and Technology in Daejeon, South Korea. This in turn confers programmability.
This approach has gained considerable momentum in the span of just a few years, with technological advances that have improved the stability and performance of Rydberg atom arrays, as well as rapid scaling from a few dozen qubits to several hundred. Early applications have focused on defined problems, such as predicting properties of materials, but the approach is versatile. “So far, any theoretical model that the theorists came up with, there was a way to implement it,” Browaeys says.
Pioneers in the field have founded companies that are developing Rydberg atom array-based systems for laboratory use, and Browaeys estimates that such quantum simulators could be commercially available in a year or two. But this work could also pave the way towards quantum computers that can be applied more generally, including in economics, logistics and encryption. Researchers are still struggling to define this still-nascent technology’s place in the computing world, but Ahn draws parallels to the Wright brothers’ early push into aviation. “That first airplane didn’t have any transportation advantages,” says Ahn, “but it eventually changed the world.”
Precise genome manipulation
For all its genome-editing prowess, CRISPR–Cas9 technology is better suited to gene inactivation than repair. That’s because although targeting the Cas9 enzyme to a genomic sequence is relatively precise, the cell’s repair of the resulting double-stranded cut is not. Mediated by a process called non-homologous end-joining, CRISPR–Cas9 repairs are often muddied by small insertions or deletions.
Most genetic diseases require gene correction rather than disruption, notes David Liu, a chemical biologist at Harvard University in Cambridge. Liu and his team have developed two promising approaches to do just that. Both exploit CRISPR’s precise targeting while also limiting Cas9’s ability to cut DNA at that site. The first, called base editing, couples a catalytically impaired form of Cas9 to an enzyme that aids chemical conversion of one nucleotide to another — for example, cytosine to thymine or adenine to guanine (see Nature https://doi.org/hc2t; 2016). But only certain base-to-base changes are currently accessible using this method. Prime editing, the team’s newer development, links Cas9 to a type of enzyme known as reverse transcriptase and uses a guide RNA that is modified to include the desired edit to the genomic sequence (see Nature 574, 464–465; 2019). Through a multistage biochemical process, these components copy the guide RNA into DNA that ultimately replaces the targeted genome sequence. Importantly, both base and prime editing cut only a single DNA strand, a safer and less disruptive process for cells.
First described in 2016, base editing is already en route to the clinic: Beam Therapeutics, founded by Liu and also based in Cambridge, got the nod in November from the US Food and Drug Administration to trial this approach in humans for the first time, with the goal of repairing the gene that causes sickle-cell disease.
Prime editing is not as far along, but improved iterations continue to emerge, and the method’s promise is clear. Hyongbum Henry Kim, a genome-editing specialist at Yonsei University College of Medicine in Seoul, and his team have shown that they can achieve up to 16% efficiency using prime editing to correct retinal gene mutations in mice12. “If we used recently reported, more advanced versions, the efficiencies would be improved even more,” he says. And Liu’s group has found that prime machinery can aid the insertion of gene-sized DNA sequences into the genome, potentially offering a safer, more tightly controlled strategy for gene therapy13. The process is relatively inefficient, but even a little repair can sometimes go a long way, Liu notes. “In some cases, it’s known that if you can replace a gene at a 10% or even a 1% level, you can rescue the disease,” he says.
Targeted genetic therapies
Nucleic acid-based medicines might be making an impact in the clinic, but they are still largely limited in terms of the tissues in which they can be applied. Most therapies require either local administration or ex vivo manipulation of cells that are harvested from and then transplanted back into a patient. One prominent exception is the liver, which filters the bloodstream and is proving to be a robust target for selective drug delivery. In this instance, intravenous — or even subcutaneous — administration can get the job done.
“Just getting delivery at all to any tissue is difficult, when you really think about the challenge,” says Daniel Anderson, a chemical engineer at the Massachusetts Institute of Technology (MIT) in Cambridge. “Our bodies are designed to use the genetic information we have, not to accept newcomers.” But researchers are making steady progress in developing strategies that can help to shepherd these drugs to specific organ systems while sparing other, non-target tissues.
Adeno-associated viruses are the vehicle of choice for many gene-therapy efforts, and animal studies have shown that careful selection of the right virus combined with tissue-specific gene promoters can achieve efficient, organ-restricted delivery14. Viruses are sometimes challenging to manufacture at scale, however, and can elicit immune responses that undermine efficacy or produce adverse events.
Lipid nanoparticles provide a non-viral alternative, and several studies published over the past few years highlight the potential to tune their specificity. For example, the selective organ targeting (SORT) approach developed by biochemist Daniel Siegwart and his colleagues at the University of Texas Southwestern Medical Center in Dallas, enables the rapid generation and screening of lipid nanoparticles to identify those that can effectively target cells in tissues such as the lung or spleen15. “That was one of the first papers that showed that if you do systematic screening of these lipid nanoparticles and start changing their compositions, you can skew the biodistribution,” says Roy van der Meel, a biomedical engineer at the Eindhoven University of Technology in the Netherlands. Several groups are also exploring how protein components such as cell-specific antibodies might assist the targeting process, Anderson notes.
Anderson is particularly excited about the preclinical progress in targeting blood and immune cell precursors in bone marrow demonstrated by companies such as Beam Therapeutics and Intellia in Cambridge, both of which are using specially designed formulations of lipid nanoparticles. Success in targeting those tissues, he says, could spare patients from the gruelling process involved with current ex vivo gene therapies, which includes chemotherapy to kill existing bone marrow before transplantation. “Doing these things in vivo could really change treatment for patients,” says Anderson.
Spatial multi-omics
The explosion in single-cell ’omics development means researchers can now routinely derive genetic, transcriptomic, epigenetic and proteomic insights from individual cells — sometimes simultaneously (see go.nature.com/3nnhooo). But single-cell techniques also sacrifice crucial information by ripping these cells out of their native environments.
In 2016, researchers led by Joakim Lundeberg at the KTH Royal Institute of Technology in Stockholm devised a strategy to overcome this problem. The team prepared slides with barcoded oligonucleotides — short strands of RNA or DNA — that can capture messenger RNA from an intact tissue slice, such that each transcript could be assigned to a particular position in the sample according to its barcode. “No one really believed that we could pull out a transcriptome-wide analysis from a tissue section,” says Lundeberg. “But it turned out to be surprisingly easy.”
The field of spatial transcriptomics has since exploded. Multiple commercial systems are now available, including the Visium Spatial Gene Expression platform from 10x Genomics, which builds on Lundeberg’s technology. Academic groups continue to develop innovative methods that can map gene expression with ever-increasing depth and spatial resolution.
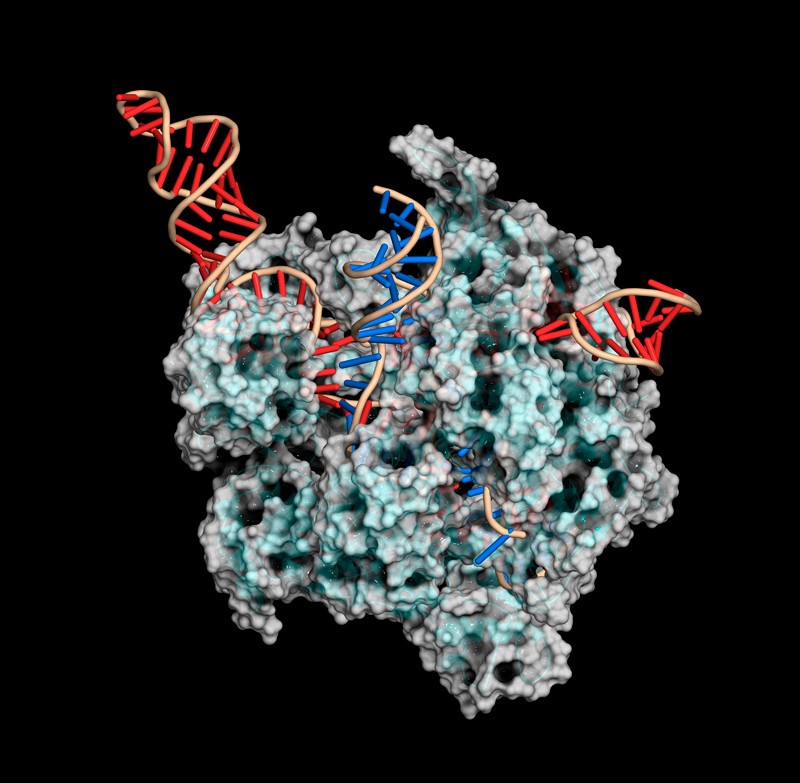
A CRISPR–Cas9 gene-editing complex uses a guide RNA (red) to cut DNA (blue).Credit: Mulekuul/SPL
Now researchers are layering further ’omic insights on top of their spatial maps. For example, biomedical engineer Rong Fan at Yale University in New Haven, Connecticut, developed a platform known as DBiT-seq16, which employs a microfluidic system that can simultaneously generate barcodes for thousands of mRNA transcripts and hundreds of proteins labelled with oligonucleotide-tagged antibodies. This can provide a much more accurate assessment of how cellular gene expression influences protein production and activity than could be obtained from transcriptomic data alone, and Fan’s team has been using it to investigate processes such as immune cell activation. “We’re seeing early signs of how immune cells in the skin react to the Moderna COVID-19 vaccine,” he says. Some commercial systems can also capture spatial data from multiple proteins in parallel with transcriptomic insights, including the Visium platform and Nanostring’s GeoMx system.
Meanwhile, Lundeberg’s group has refined its spatial transcriptomics method to simultaneously capture DNA sequence data. This has allowed his team to start mapping the spatiotemporal events underlying tumorigenesis. “We could follow these genetic changes in space, how they evolve into additional genetic variants that eventually lead to the tumour,” he says.
Fan’s team has demonstrated spatial mapping of chromatin modifications in tissue samples, which can reveal the cellular gene regulatory landscapes that influence processes such as development, differentiation and intercellular communication17. Fan is confident that the method can be paired with spatial analysis of RNA, and even proteins. “We have preliminary data showing that this is totally doable,” he says.
CRISPR-based diagnostics
The CRISPR–Cas system’s capacity for precise cleavage of specific nucleic acid sequences stems from its role as a bacterial ‘immune system’ against viral infection. This link inspired early adopters of the technology to contemplate the system’s applicability to viral diagnostics. “It just makes a lot of sense to use what they’re designed for in nature,” says Pardis Sabeti, a geneticist at the Broad Institute of MIT and Harvard in Cambridge. “You have billions of years of evolution on your side.”
But not all Cas enzymes are created equal. Cas9 is the go-to enzyme for CRISPR-based genome manipulation, but much of the work in CRISPR-based diagnostics has employed the family of RNA-targeting molecules known as Cas13, first identified in 2016 by molecular biologist Feng Zhang and his team at the Broad. “Cas13 uses its RNA guide to recognize an RNA target by base-pairing, and activates a ribonuclease activity that can be harnessed as a diagnostic tool by using a reporter RNA,” explains Jennifer Doudna at the University of California, Berkeley, who shared the 2020 Nobel Prize in Chemistry with Emmanuelle Charpentier, now at the Max Planck Unit for the Science of Pathogens in Berlin, for developing the genome-editing capabilities of CRISPR–Cas9. This is because Cas13 doesn’t just cut the RNA targeted by the guide RNA, it also performs ‘collateral cleavage’ on any other nearby RNA molecules. Many Cas13-based diagnostics use a reporter RNA that tethers a fluorescent tag to a quencher molecule that inhibits that fluorescence. When Cas13 is activated after recognizing viral RNA, it cuts the reporter and releases the fluorescent tag from the quencher, generating a detectable signal. Some viruses leave a strong enough signature that detection can be achieved without amplification, simplifying point-of-care diagnostics. For example, last January, Doudna and Melanie Ott at the Gladstone Institute of Virology in San Francisco, California, demonstrated a rapid, nasal-swab-based CRISPR–Cas13 test for amplification-free detection of SARS-CoV-2 using a mobile phone camera18.
RNA-amplification procedures can boost sensitivity for trace viral sequences, and Sabeti and her colleagues have developed a microfluidic system that screens for multiple pathogens in parallel using amplified genetic material from just a few microlitres of sample19. “Right now, we have an assay to do 21 viruses simultaneously for less than US$10 a sample,” she says. Sabeti and her colleagues have developed tools for CRISPR-based detection of more than 169 human viruses at once, she adds.
Other Cas enzymes could flesh out the diagnostic toolbox, Doudna notes, including the Cas12 proteins, which exhibit similar properties to Cas13 but target DNA rather than RNA. Collectively, these could detect a broader range of pathogens, or even enable efficient diagnosis of other non-infectious diseases. “That could be very useful if you could do that relatively quickly, especially as different cancer subtypes become defined by particular types of mutations,” Doudna says.

